






01
PROTAC的历史发展

图1. PROTAC技术的原理——泛素介导的蛋白降解途径,来源:Swatek, Kirby N, and David Komander. “Ubiquitin modifications.” Cell research vol. 26,4 (2016): 399-422.
2004年的诺贝尔化学奖授予了发现泛素调节的蛋白降解途径的三位科学家,这是一种泛素-蛋白酶体系统(UPS)参与的蛋白降解途径。首先通过消耗1分子ATP激活泛素小分子,使它与一种泛素激活酶E1形成复合物,随后这个泛素分子又转移到了泛素结合酶E2上形成复合物,当泛素连接酶E3识别结合底物蛋白时,这个复合物会紧接着和它结合,泛素就会直接或者间接地从E2转移到底物蛋白上。
泛素小分子的羧基端会和底物蛋白的Lys上的赖氨酸残基共价结合,当这个泛素传递的过程重复发生多次后,泛素根据其自身不同的结合位点以一些特定的方式聚集,就会使这个底物蛋白被蛋白酶体降解,随后通过去泛素化作用,这些泛素分子可以被循环利用。泛素-蛋白酶体系统是细胞内蛋白质降解的主要途径,参与细胞内80%以上的蛋白降解过程。
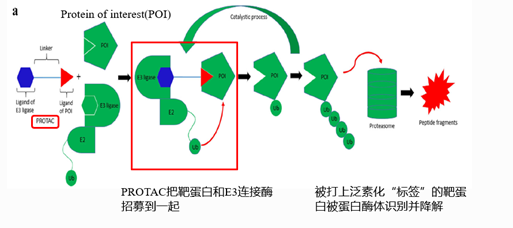
图2. PROTAC技术的基本原理,来源:Li, Xin, and Yongcheng Song. “Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy.” Journal of hematology & oncology vol. 13,1 50. 13 May. 2020
本文所介绍的PROTAC这项技术,全称是PROteolysis-Targeting Chimeras,即靶向蛋白水解的嵌合体,它的原理就是依赖于这条泛素化的蛋白降解途径。
在结构上,PROTAC包括三个部分:一个E3泛素连接酶配体和一个靶蛋白配体,两个活性配体通过特殊设计的“Linker”结构连接在一起,最终形成了三联体的“PROTAC”的活性形式,这样的化学分子既可以结合E3泛素连接酶,又可以结合胞内蛋白质,它相当于一把“双向抓手”,通过把靶向的蛋白质招募到E3泛素连接酶附近来实现靶向蛋白质的多泛素化,最后被蛋白酶体识别和降解。
在这个过程中,PROTAC可以循环使用,因为它本身没有跟泛素结合的氨基酸位点,所以不被蛋白酶体降解。
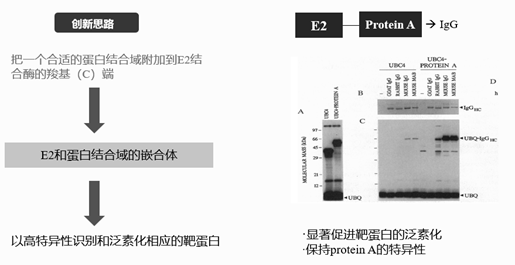
图3. PROTAC研究历史——1995年报道第一个人工诱导的蛋白降解,来源:Gosink, M M, and R D Vierstra. “Redirecting the specificity of ubiquitination by modifying ubiquitin-conjugating enzymes.” PNAS vol. 92,20 (1995): 9117-21
这种人工诱导的蛋白降解最早报道是在1995年,研究者设计出了一系列可以与IgG结合嵌合的E2结合酶,他们的创新思路是把一个蛋白结合域附加到E2结合酶的C端,从而形成一个含有靶蛋白结合域的E2嵌合体,这个嵌合体就会基于自身的性质去特异性识别并且泛素化相应的靶蛋白。
他们选择了一种E2结合酶(UBC4)和protein A嵌合,protein A可以特异性识别多种IgG抗体。这种构造的E2嵌合体具有很好的原始E2的活性,可以结合泛素。它能够显著促进靶蛋白IgG的泛素化,这种E2嵌合体的特异性与protein A一致,由于proteinA能够识别鼠和兔的IgG而不识别山羊的IgG,因此该嵌合体也能够识别鼠和兔的IgG而不识别山羊的IgG。而且这些人工诱导的泛素化特征是可以被蛋白酶体识别降解的。这项基于E2嵌合体的创新研究,首次实现人工诱导蛋白降解,为PROTAC技术奠定了基础。
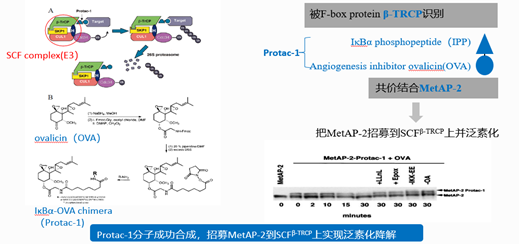
图4. PROTAC研究历史——2001年提出PROTAC概念并设计出首个PROTAC分子,来源:Sakamoto, K M et al. “Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation.” PNAS vol. 98,15 (2001): 8554-9.
2001年,耶鲁大学的Craig Crews教授与加州理工学院的Ray Deshaies教授联手发表了一篇具有里程碑意义的论文,他们利用含SCFβ-TRCP的E3诱导了MetAp-2的降解,首次引入了靶向嵌合蛋白水解(PROTAC)一词,他们将他们设计的PROTAC分子命名为PROTAC-1,它的一端是E3连接酶的配体,IκBα 磷酸肽,另一端是靶蛋白MetAP-2配体,血管生成抑制剂ovalicin,利用这把“双向抓手”,把靶蛋白MetAP-2招募到SCFβ-TRCP上,实现泛素化和蛋白酶体降解。
他们成功合成出了这样的PROTAC-1分子,并且证实了它降解靶蛋白的能力。当在体系中加入了LLnL(一种蛋白酶体抑制剂时),降解能力受到抑制,证明了这个PROTAC-1分子确实是依赖蛋白酶体催化靶蛋白的降解。但是PROTAC-1作为第一代PROTAC分子也表现出一个显著的缺点,它含有一个分子量比较大的肽类的配体,从而限制了它的细胞膜渗透性,同时它也不具有很好的稳定性,细胞活性低,因此限制了它的可应用性,无法应用到药物开发中。
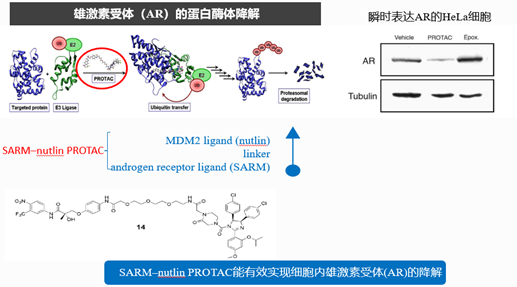
图5. PROTAC研究历史——2008年报道首个全小分子PROTAC,来源:Schneekloth, Ashley R et al. “Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics.” Bioorganic & medicinal chemistry letters vol. 18,22 (2008): 5904-8.
2008年,这个含有肽的问题得到了解决,报道了第一个全小分子的PROTAC。这项研究中合成了能够靶向降解雄激素受体(AR)的SARM-nutlin PROTAC小分子,其中Nutlin是一种咪唑啉衍生物,研究中发现它可以与E3连接酶MDM2的天然底物p53竞争性结合MDM2,因此可以作为E3连接酶配体。
在细胞实验中,将其作用于瞬时表达AR的HeLa细胞中,发现这个小分子表现出比较好的细胞渗透性、稳定性和一定的降解能力,但其细胞活性仍不理想,小分子在10μM浓度下才能有效诱导AR降解,因此仍然限制了它的成药性。
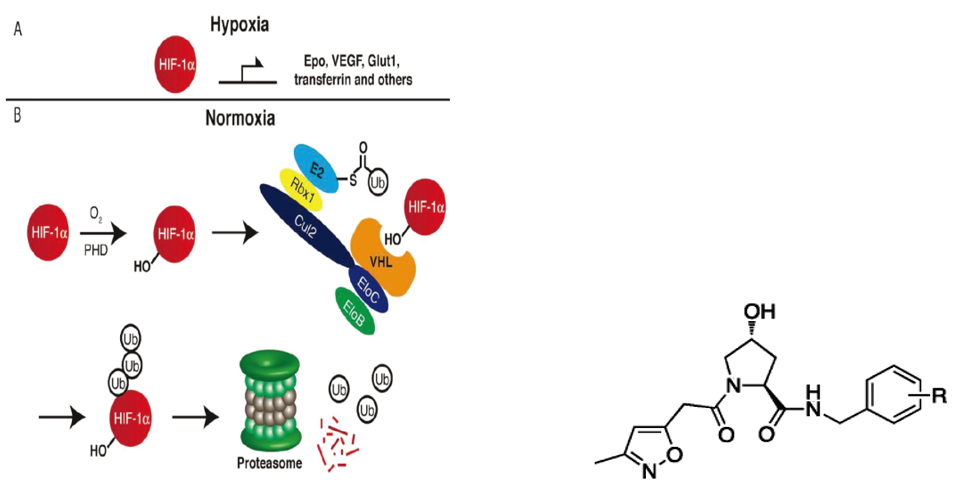
图6. HIF-1α与VHL蛋白相互作用(左);一类具有高亲和力的VHL配体(右),来源:Buckley, Dennis L et al. “Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction.” Journal of the American Chemical Society vol. 134,10 (2012): 4465-8.
2010-2012年,随着Craig Crews团队和Alessio Ciulli及其他合作者发现了更多泛素E3连接酶的结合体,在类药E3结合配体的发现与优化方面,PROTAC取得了重大的进展。
例如发现了methyle bestatin(MeBS)可以作为IAP E3连接酶配体,产生了IAP-based PROTAC;
还发现沙利度胺(thalidomide)这个明星药物可以作为CRBN E3连接酶配体,产生了CRBN-based PROTAC。
2012年,VHL的高亲和力拟肽配体被发现,HIF-1α(缺氧诱导因子)是一种转录因子,在缺氧条件下被激活,上调促红细胞生成素,血管内皮生长因子,葡萄糖转运蛋白,转铁蛋白等的表达。它可以与E3连接酶中的VHL相互作用,被泛素化并进入蛋白酶体降解途径。对这条降解途径的抑制可以有效替代慢性贫血等由于缺乏HIF-1α引起的疾病治疗药物。
结合以前旨在抑制这条途径展开的VHL抑制剂小分子的设计研究,研究者开展了对于VHL配体的筛选和优化,获得了具有高亲和力的配体,设计出了VHL-based PROTAC分子。(右图分子式为一类优化的VHL配体)
这种把VHL的抑制剂小分子研究应用到PROTAC设计中的组合理念,促成了Craig Crews教授创办生物技术公司Arvinas,该公司专注于PROTAC技术,开发创新药物,使得PROTAC技术得以迅速地发展。Arvinas基于VHL配体设计的PROTAC化合物具有更多类药特性,可以大幅度提高细胞的穿透性,细胞活性可以达到纳摩尔级别。
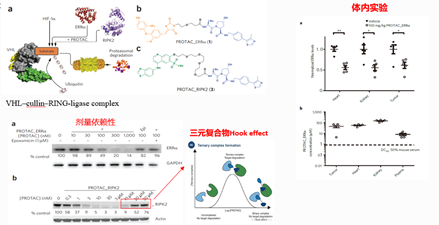
图8. PROTAC研究历史——PROTAC分子在体内实现催化下调蛋白水平,来源:Bondeson, Daniel P et al. “Catalytic in vivo protein knockdown by small-molecule PROTACs.” Nature chemical biology vol. 11,8 (2015): 611-7.
2015年这个团队发表了一项重要研究,他们设计了两种基于VHL E3 ligase的小分子PROTAC,一种带有雌激素相关受体(ERRα)的配体,另一种是RIPK1蛋白激酶的配体,中间通过linker与高亲和性的VHL配体连接,靶蛋白的水平与癌症、自身免疫性疾病发病有关的。
这两个小分子PROTAC都表现出剂量依赖性,但是当PTORAC小分子浓度过高时,会出现三元复合物中常见的hook effect,降解靶蛋白的能力反而下降。这是因为当PROTAC小分子浓度过高时,以形成二元复合物为主,而三元复合物的形成受到了抑制(如上图)。
这项研究还证实了这样的小分子PROTAC可以通过注射到小鼠体内,进入不同部位,然后降低其体内不同器官中的相应靶蛋白水平。从而说明PROTAC能够剂量依赖性地下调靶蛋白的水平,并且在小鼠体内有效发挥作用。
有了这些研究的基础,利用PROTAC技术进行小分子药物开发有了广阔的前景,但是PROTAC技术具体是怎么设计元件的,以及如何突破许多瓶颈在体内工作?
02
PROTAC设计与工作
简单回顾一下PROTAC的工作原理:由linker连接的两个配体分别靶向目标蛋白(POI)和酶E3,拉近两者距离,以促进泛素转移,从而使POI跳过常规UPS步骤,“半路出家”走向被蛋白酶体识别、降解的不归路。箭头箭柄和两个靶标,互相靠近后,产生化学反应,磨擦出火花——不难看出,PROTAC似乎扮演“丘比特之箭”的角色,尽管这是一支“分手箭”。
丘比特之箭的一生可分为四个阶段:
铸箭,巧妇难为无米之炊,一支恰到好处的箭是爱神降临的资本;
瞄准,乱点鸳鸯谱将会酿成悲剧;
射箭,神仙眷侣的相逢也需要天时地利;
箭效,色厉内荏还是内外兼修,只有正确引导结局走向的箭才拥有圆满一生。
相应地,PROTAC的设计与工作历程也可以“四步走”,即对PROTAC的量与质的设计、PROTAC靶向性方面的特点、提高PROTAC可控性的策略以及对PROTAC执行力的评估。
1
铸箭——PROTAC设计
PROTAC的自我修养有哪些?简单来说,合适配体和合适连接体是其达到及格线的第一步。
配体选择需因物制宜:对POI保证亲和性,对E3还需格外注意其泛素转移酶活性的保留。连接体则要“扬长避短”,尽可能降低对药物入膜的不利影响,同时稳定三元复合物,达到“不惹事也不怕事”的效果。
目前PROTAC设计日臻完善(图9),干实验与湿实验结合,借助计算机模拟、药效实验数据以及复合物结构信息有效地挖掘小分子化合物库,并不断优化药物。
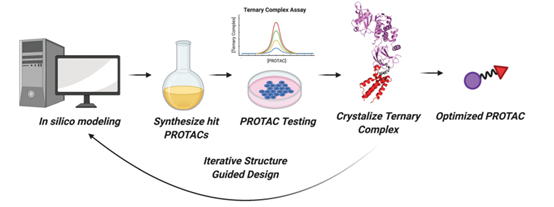
图9. PROTAC研发流程图,来源:Bond, Michael J, and Craig M Crews. “Proteolysis targeting chimeras (PROTACs) come of age: entering the third decade of targeted protein degradation.” RSC chemical biology vol. 2,3 725-742. 19 Mar. 2021, doi:10.1039/d1cb00011j
其中配体设计与筛选始终是研究热点。配体类别上,除了传统的经改良的小分子化合物,核酸分子也已被开发为POI配体,在靶向转录因子方面表现不俗(图10)。
配体靶标相互作用上,一方面是相互作用方式,配体能直接或间接结合靶标,比如借助囊泡膜蛋白这一桥梁,配体与囊泡结合,囊泡则靶向POI,从而拉近POI与E3。另一方面则是相互作用强弱。相互作用强意味着三元复合物十分稳定,PROTAC对靶标“死心塌地”,甚至“殉情”,沦为“一次性”药物。相互作用弱虽然提高复合物不稳定的风险,但PROTAC可以“快刀斩乱麻”,快速抽身而投入下一轮降解,提高整体降解速率。面临向稳还是向快的岔路口,近些年来崭露头角的可逆共价PROTAC或许能够另辟蹊径。
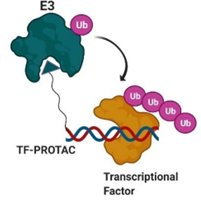
图10. 靶向转录因子的PROTAC工作示意图,蓝红螺旋即为核酸分子,与转录因子直接结合从而拉近E3与POI,图片来源:Hu, Zhenyi, and Craig M Crews. “Recent Developments in PROTAC-Mediated Protein Degradation: From Bench to Clinic.” Chembiochem : a European journal of chemical biology, 10.1002/cbic.202100270. 8 Sep. 2021, doi:10.1002/cbic.202100270

图11. 两种高效筛选配体的方法工作示意图。左图为chemo proteomic competition,右图为DNA encoded library。图片来源:Bond, Michael J, and Craig M Crews. “Proteolysis targeting chimeras (PROTACs) come of age: entering the third decade of targeted protein degradation.” RSC chemical biology vol. 2,3 725-742. 19 Mar. 2021, doi:10.1039/d1cb00011j
方法论上,近年出现两种配体高效筛选的策略(图3)。一是chemo proteomic competition,它是指对细胞或机体所有蛋白质,设置仅有待测配体和待测配体与已知共价配体竞争性结合的两个实验组,通过比较与兴趣蛋白结合的两种配体的比例评估待测配体与蛋白质的亲和力。
二是DNA encoded library,即将小分子化合物与DNA连接而对其进行“DNA编码”后再与靶标蛋白混合进行亲和筛选,最后通过DNA序列按图索骥,锁定潜在配体。通过工作原理可以初步推断,两者分别侧重于筛选共价结合配体和非共价结合配体。
PROTAC的元件及其品质初步达标后,接下来要考虑的问题是,打造多少支医药丘比特之箭恰到好处?由于三元复合物往往呈现钟形动力学曲线,所以PROTAC与有效复合物并不呈现简单的正相关关系。
此外,某些PROTAC形成三元复合物时具有正协同效应,加之可循环PROTAC的“催化”特性,低剂量高疗效成为PROTAC的一块金字招牌。总之,PROTAC设计的质与量,在精不在多。
2
瞄准——PROTAC靶向性
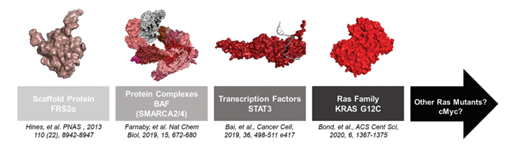
图12. PROTAC靶向的POI发展时间线,来源:Bond, Michael J, and Craig M Crews. “Proteolysis targeting chimeras (PROTACs) come of age: entering the third decade of targeted protein degradation.” RSC chemical biology vol. 2,3 725-742. 19 Mar. 2021, doi:10.1039/d1cb00011j
目前PROTAC已被证明能够有效降解支架蛋白、蛋白复合体、转录因子和undruggable蛋白这四类被普遍认为缺少有效结合小分子药物位点的POI(图4)。相较于POI“百花齐放”的喜人之势,VHL、CRBN等从发展初期就被使用的酶几乎垄断了E3连接酶选择。
有趣的是PROTAC似乎是一支不锋利的神箭,低亲和力配体也能以较高选择性靶向目标。这种现象可能是由于“精心设计”的linker对三元复合物起额外的稳定作用,发挥了上文提到的“不怕事”作用。换言之,只要配体自身实力尚可,有强大背景加持仍能“出道”——不过这仍需要精妙的PROTAC设计为基础。
3
射箭——PROTAC可控性
神箭之外,天时地利也格外重要。
首先我们要借助外源信号让PROTAC“伺机而动”,定时起效。一种策略是光控制,它又可分为“光笼法”和“光开关”两种方法(图5)。
前者是在PROTAC的配体上添加“光笼基团”以钝化正常状态下的PROTAC,经特定波长光照射后“光笼基团”脱落,实现不可逆的PROTAC启动。
后者则是在PROTAC的linker中引入偶氮苯基团,切换不同波长的光会改变linker的顺反构型,而只有一种构型有利于三元复合物形成,这样便形成可逆PROTAC开关。
胞外信号分子也是一个有力把手。比如筛选出特定结合位点,使得只有磷酸化的PROTAC才能与POI紧密结合,这样便能够通过定时添加生长因子,经系列信号转导使PROTAC打上磷酸化标签,作为其“射箭资格证”。
然而,PROTAC的时间可控性面临药物透膜性变差、光辐射诱变性以及组织特异性差等问题。
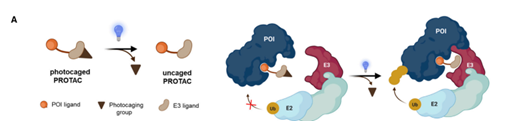
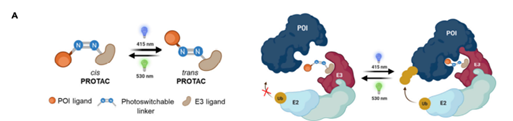
图13. 光控法示意图。上图为“光笼法”,橙色圆球即为光笼基团。下图为“光开关”,蓝色部分即为引入的偶氮苯基团。图片来源:Bond, Michael J, and Craig M Crews. “Proteolysis targeting chimeras (PROTACs) come of age: entering the third decade of targeted protein degradation.” RSC chemical biology vol. 2,3 725-742. 19 Mar. 2021, doi:10.1039/d1cb00011j
组织特异性问题恰恰与“地利”相对应,即确保PROTAC在目标组织或细胞发挥作用。形象地说,PROTAC和细胞犹如列车和乘客,对PROTAC的空间性控制则如同防范乘客逃票,一要抓上车前检票,二要抓上车后查票。
“检票”,使持有特定marker的细胞摄入PROTAC。比如以肿瘤细胞高表达的叶酸受体为“票据”,使与叶酸结合的PROTAC只能进入肿瘤细胞。再比如利用抗原-抗体特异性结合作用,将PROTAC与抗体绑定,靶向特定细胞。
“查票”,其原理是非目标细胞即便摄取PROTAC,但由于胞内缺少特定E3或蛋白酶体不识别复合物等启动降解的必要条件,无法搭乘PROTAC这班“死亡列车”。
4
箭效——PROTAC执行力
医药丘比特之箭究竟箭效如何?理论上,PROTAC是典型的药物差等生,在“类药五原则”的及格线外挣扎,且其ADME尚未明朗,很难评估其生物利用度。
实验数据却显示PROTAC是一支潜力股。借助各类技术手段从POI降解效果、PROTAC主体地位验证以及毒副反应检测三个角度对PROTAC进行评估后,我们往往能够惊喜地发现,PROTAC疗效高于理论预期。
03
PROTAC的优点与缺点
1
PROTAC的优缺点
首先第一个优点是作用范围更广、活性更高、可靶向“不可成药”靶点。传统的小分子和抗体等都是通过“占据驱动”的作用模式抑制靶蛋白的功能发挥治疗疾病的作用,这种作用模式需要抑制剂或单抗具备较高的浓度才能够占据靶点的活性位点,阻断下游信号通路的转导。然而,据估计,人类细胞中80%的蛋白缺乏这样的位点。
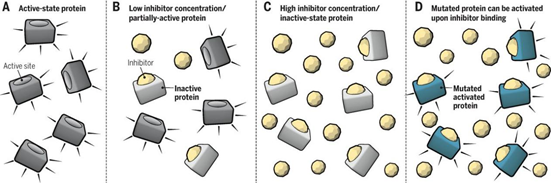
图14. 传统小分子的“占据驱动”,来源:Science. 2017, 355(6330): 1163-1167.
PROTAC只是提供结合活性,触发靶蛋白与E3酶结合从而引发降解这一事件,属于“事件驱动(event driven)”,不需要直接抑制目标蛋白的功能活性,药物不需要与目标蛋白长时间和高强度的结合,因此可以靶向传统难以成药的蛋白(undruggable target)由于PROTAC只需要与目标蛋白弱结合就可以特异性地“标记”它,因此,目前蛋白质组中80%不可成药的蛋白可能都能够用PROTAC技术来解决。
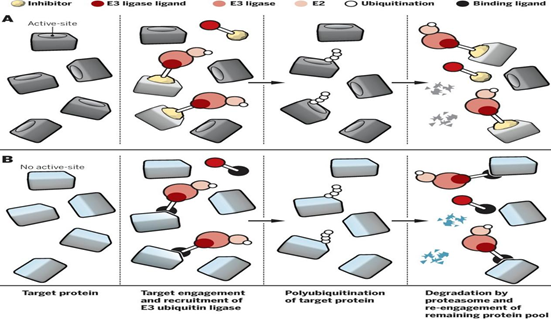
图15. PROTAC的“事件驱动”。Science. 2017, 355(6330): 1163-1167.
据不完全统计,目前已经有超过100种蛋白被成功降解。这些靶点包括:
(1)激酶类,如RIPK2、BCR-ABL、EGFR、HER2、c-Met、TBK1、CDK2/4/6/9、ALK、Akt、CK2、ERK1/2、FLT3、PI3K、BTK、Fak等;
(2)BET蛋白,如BRD2/4/6/9;
(3)核受体,如AR、ER等;
(4)其他蛋白,如MetAp-2、Bcl-xL、Sirt2、HDAC6、Pirin、SMAD3、ARNT、PCAF/GCN5、Tau、FRS2等。
这其中就包括“不可成药靶点”,如转录因子调节蛋白pirin、表观遗传相关蛋白PCAF/GCN5等。另外,根据Nature 3月份的一项报告显示,到2021年年底,将至少有15款蛋白降解剂(包含PROTAC和分子胶)进入临床试验。
讲到不可成药靶点,RAS(KRAS、HRAS和NRAS)肯定是绕不要过去的。作为癌症中最常见的突变基因,RAS是肺癌、结直肠癌和胰腺癌的重要驱动因素。被发现和研究40多年来,一直没有针对该靶点的药物上市。终于在今年5月份,Amgen公司开发的KRAS-G12C不可逆抑制剂AMG510(Sotorasib)成功上市,总算是终结了该靶点的“不可成药性”。
虽然KRAS的不可成药性被小分子抑制剂终结了,但是降解剂在该靶点仍然大有可为。针对KRAS-G12C突变,Crews等人在KRAS抑制剂AMG510和MRTX849的基础上,设计并合成了相关的PROTAC,活性测定发现了具有良好降解活性的降解剂,这或许可以为进一步攻克RAS提供新的解决方案。
可以预见,利用PROTAC技术攻克的可能不只是G12C,对其他突变或许也能有所作为。
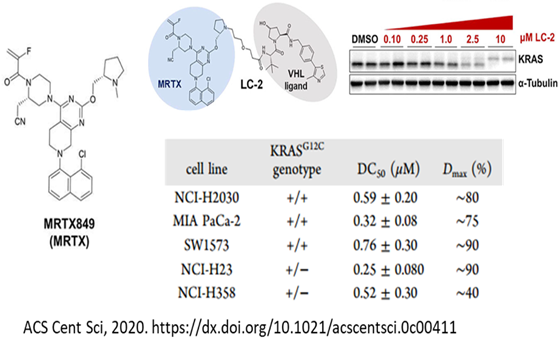
图16. 针对RAS靶点的相关PROTAC药物活性测定
第二个优点是PROTAC在某些靶点上可实现小分子难以实现的选择性。多靶点酪氨酸激酶抑制剂Foretinib可以结合130多种激酶,Crews等人将其作为结合靶蛋白的配体,分别连接E3连接酶VHL(von Hippel-Lindau)和CRBN(cereblon)的配体得到相应的PROTAC,结果显示连接VHL和CRBN的PROTAC只能分别降解36和62种蛋白,而只有12种蛋白才能被这两种PROTAC降解。
2,4-二氨基嘧啶骨架是激酶抑制剂的常见骨架,EGFR、ALK、CDK、Jak等激酶的抑制剂都有用到其作为母核。但是该骨架形成的药物分子大多都比较“脏”,也就是靶点选择性比较差,经常对其他很多激酶都有很强的抑制活性,开发过程中off target的脱靶效应常常成为毒副作用的主要来源,影响新药开发的成功率。
Gray课题组基于该骨架合成的PROTAC虽然能够结合190多种激酶,但是细胞实验中只能降解12和22种激酶,大大提高了靶点的选择性。
PROTAC技术第三个优点是不仅可以实现小分子抑制剂难以做到的选择性,在提高活性方面也具有非常显著的优势。以PROTAC技术应用最早的靶点BET(BRD2/3/4)为例,秦冲等人发现的QCA570在细胞抗增殖活性方面表现出了明显优于JQ1等抑制剂的能力,JQ1等抑制剂的活性大都在纳摩尔级水平,而QCA570的活性提高了三个数量级,达到了惊人的皮摩尔级水平。基于出色的体外细胞抗增殖活性,降解剂在体内也表现出了强效抗肿瘤活性,而且呈现出给药剂量低和给药频次也低的特点。这说明在类似BET等靶点中,PROTAC具有明显的优势。
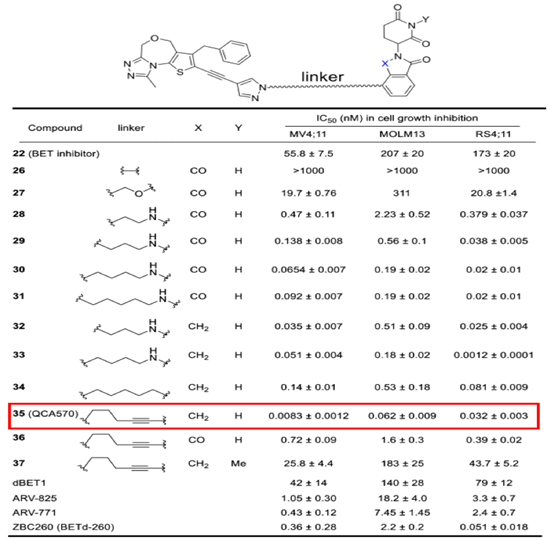
图17. PROTAC可以提高活性和安全性,来源:J Med Chem 2018, 61, 6685-6704.
第四个优点是克服药物的耐药性。小分子抑制剂或拮抗剂在临床用药过程中,不可避免的都会发生获得性耐药。比如EGFR-T790M和C797S耐药等。虽然可以通过开发新一代的抑制剂,如第三代和第四代EGFR来解决耐药问题,但是随着新一代药物的使用,新的耐药也会随之出现。PROTAC技术在克服耐药方面已经显现出了一定的优势。
Crews、Jian Jin、张三奇、丁克和Gray等课题组都有相关EGFR-PROTACs的研究,旨在通过蛋白降解途径克服耐药突变或找寻突破抑制剂的蛋白降解疗法。可以看出,第一、二、三、四代抑制剂作为结合靶蛋白EGFR的配体,都有被应用到PROTAC的设计中。其中,Gray课题组将变构抑制剂也应用到了PROTAC当中,并取得了不错的效果,能够选择性的降解不同的EGFR突变体,而规避对野生型的降解。体外抗细胞增殖活性也进一步验证了这种选择性。
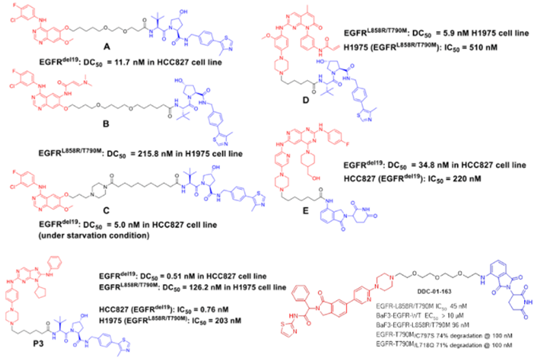
图18. EGFR-PROTACs. [1] Cell Chem Biol 2018, 25, 67-77. [2] J Med Chem 2020, 63, 1216-1232. [3] Eur J Med Chem 2020, 189, 112061. [4] Eur J Med Chem 2020, 208, 112781. [5] Eur J Med Chem 2020, 192, 112199. [6] Angew Chem Int Ed 2020, 59, 14481.
下表是PROTAC与其他几种药物的特征对比,可以看出PROTAC在多个方面都比已有的三种药物更加优异。除此之外,PROTAC还有其他优点:
(1)用量小,催化剂量即可;
(2)清除蛋白堆积;
(3)毒性低;
(4)不依赖于亲和力,可选择性高;
(5)克服靶蛋白突变/过表达引起的耐药等。
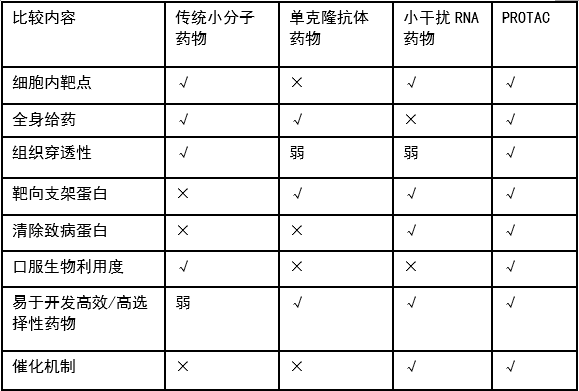
表1. PROTAC药物与其他几种药物的特征对比
2
PROTAC存在的缺点和挑战
首先是三联体成药性差。传统的PK、PD模型可能不适用于PROTAC技术,PROTAC的催化特性以及相对复杂的三联体结构,可能使得组织分布不遵循传统的pH分布理论,还有可能出现靶点介导的药物处置现象导致PK/PD复杂化,但因目前相关的体内试验较少,还没有建立出针对PROTAC技术应用的的PK、PD模型。
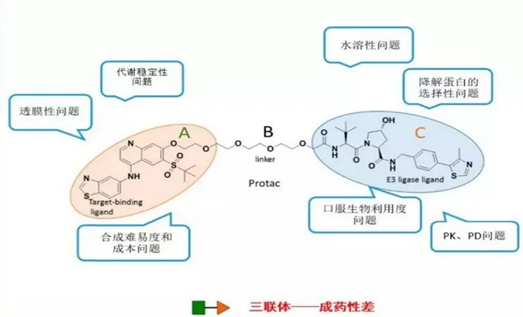
图19. PROTAC三联体成药性差,图源:http://new.qq.com/omn/20210322/20210322A0EICO00
对于传统的小分子药物,可以用成药性五原则对其进行初步的判断,但对于PROTAC来讲这一规律已经不能适用。PROTAC相当于是双靶点药物,分子量相对一般的小分子药物较大,在700道尔顿以上,有的甚至超过1000道尔顿。这可能会影响到分子的水溶性及透膜性,不利于采用口服方式给药的方式。目前常用的方式是经腹膜内,皮下或静脉内注射方式给药。
第三个缺点是三元复合体形成困难。PROTAC发挥药效必须与目标靶蛋白和E3连接酶结合形成三元复合物。PROTAC分子太多或太少都不利于三元复合体的形成。成功形成三元复合体首先需要成功通过细胞膜进入细胞,然后控制浓度以避免药物分子与靶蛋白和E3连接酶分别形成二元复合物(hook effect,钩效应),最后还要充分考虑靶蛋白与E3连接酶接触区域的电荷排斥和立体结构排斥。

图20. PROTAC三元复合体形成困难,来源:http://new.qq.com/omn/20210322/20210322A0EICO00
与设计和定制siRNA或RNA相比,PROTAC开发周期相对较长。如下图所示,PROTAC不仅需要蛋白质的配体,还需要蛋白质转化成PROTAC,这可能是一个耗时的过程,涉及大量的合成化学及药物化学相关支持。基于标记的系统(HaloPROTACs和dTAG)虽然绕过了发现阶段,但也同时废除了PROTAC系统在可移植性(无需对其基因进行修饰)方面的优势。
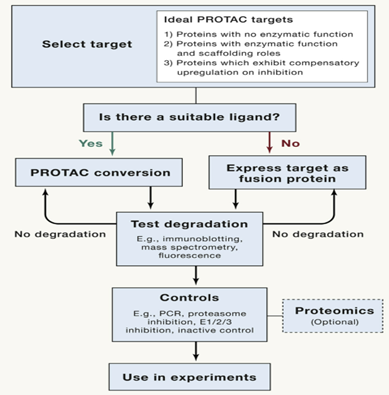
图21. PROTAC的开发周期,来源:http://new.qq.com/omn/20210322/20210322A0EICO00
除以上内容,PROTAC还存在一些其他缺点与挑战:
(1)Linker的选取问题;
(2)靶蛋白降解困难;
(3)药效性来源有待商榷;
(4)可用的E3泛素连接酶数量有限;
(5)PROTAC技术并非适用所有的蛋白质或亚细胞位置等。
04
PROTAC的应用与前景
先讲讲它的应用,前文讲过了PROTAC从1995年到2015年的发展历程,也是一步步的向着成药性在发展。尤其是在2013年,[kruːz]建立了Arvinas之后,它在2015年首次被带入了药物应用。而本部分的重点部分是AVS-110,AVS-378这两个PROTAC分子。
在2001年,[kruːz]等人合理地利用与泛素连接酶E3体系SCF结合的10肽,加上甲硫氨酰氨肽酶2的共价抑制剂,设计并合成了首个用于降解MetAP2的PROTAC-1。蛋白试验验证了PROTAC-1可快速降解MetAP2,且它可以与那个共价抑制剂和10肽竞争性结合MetAP2和SCF。
但是这个开创性的工作并没有就此结束,研究者提出了更进一步的问题。利用SCF的E3体系,是否可以设计不同的PROTAC用于降解其他致病蛋白?共价键蛋白抑制剂可以设计PROTAC,非共价键的天然底物能否用于设计PROTAC?PROTAC在蛋白水平试验有效,在细胞内是否也能产生同样的降解作用?
带着这些问题,他们选择了与前列腺癌相关的雄激素受体(AR)和与乳腺癌相关的雌激素受体(ER)作为降解的目标蛋白。很有意思是,这两个靶蛋白成为了后来Arvinas公司也是目前全球推进最快的PROTAC降解的蛋白。这两张图就是将AR和ER泛素化的示意图。
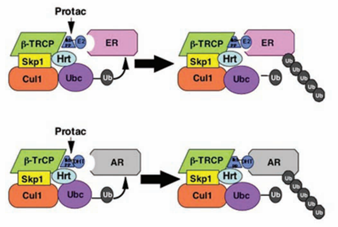
图22. AR和ER泛素化的示意图,来源:Kathleen M. Sakamoto, Kyung B. Kim, Rati Verma, Andy Ransick, Bernd Stein, Craig M. Crews, Raymond J.Deshaies,Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation
那么让我们先来看一下做这两种药的重要性。以前列腺癌为例,为什么我们说要给AR泛素化,去治疗前列腺癌?
那我们先来看看目前治疗前列腺癌的方式:使用雄激素合成抑制剂阿比特龙和雄激素受体阻滞剂恩杂鲁胺联合治疗,可以将疾病早期的未化疗耐雄激素限制疗法的前列腺癌患者中位总体生存期提高到53.6个月,另外提一下,被誉为癌症治疗新希望的免疫疗法目前对耐雄激素限制疗法的前列腺癌一点办法也没有,而且一般的前列腺癌都会演变成这种,患者平均中位生存时间仅12.3个月。但遗憾的是2012年获FDA批准,到现在还没有进口。
为了降解ER和AR,研究者分别利用了其天然底物雌二醇(E2)和二氢睾脂酮(DHT),通过中间的Linker连接IκBα磷酸化的10肽,设计了PROTAC-2和PROTAC-3。这次用来设计PROTAC的小分子为天然底物的激动剂,而非抑制剂,这说明设计PROTAC不一定非要选择抑制剂,激动剂也是可以的。
接下来是前景部分。这里展示了8种常用E3连接酶以及超20个抗癌靶点,其前景不言而喻。只是目前来看,PROTAC虽然具有治疗多种疾病的前景,但目前这类药物主要被调查用于治疗癌症。



表2. 小分子PROTAC成功靶向的关键癌症靶点,表格来源:Khan S, He Y, Zhang X, Yuan Y, Pu S, Kong Q, Zheng G, Zhou D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics.
PROTAC技术给我们的启发就是,以后成药不一定非要去找一个蛋白的抑制靶点,可以找这个蛋白随意的一个靶点,只要足够特异,不会引起off target毒性就行,然后把这个靶点的配体放在哑铃的一端,就可以成药。
对于刚才讲的XOR抑制也是如此,别嘌醇和非布司他都是XOR的抑制剂,可以只去找它的结合位点,不一定要抑制它,那就能做出一个几乎没有副作用的药。
